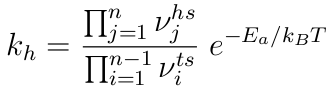- Περιγράψτε σύντομα τις φυσικές προσεγγίσεις που εμπεριέχονται σε έναν υπολογισμό με το πρόγραμμα dacapo, ακολουθώντας τα βήματα: 1) πραγματικό υλικό 2) εξίσωση Schrodinger για ηλεκτρόνια 3) εξίσωση Schrodinger για ένα ηλεκτρόνιο 4) μελέτη μόνο ηλεκτρονίων σθένους 5) δυναμικό ανταλλαγής και συσχέτισης.
- Ακολουθήστε το παράδειγμα για το μόριο CO.
Αντιγράψτε το αρχείο co.py και τρέξτε το με την εντολή
qsub co.py
Δείτε τα αποτελέσματα στο αρχείο με επέκταση .txt. Στη συνέχεια, δώστε την εντολήag CO.nc
Τι παρατηρείτε; Εξετάστε πώς αλλάζει η ενέργεια του CO καθώς αλλάζει το cell. Πόσο μεγάλο κουτί απαιτείται για να έχετε την ενέργεια με ακρίβεια πρώτου δεκαδικού ψηφίου; Τι θα άλλαζε αν το μόριο δεν ήταν πολικό (πχ O2); - Εξηγείστε τι είναι η παράμετρος planewavecutoff. Επαναλάβετε τον παραπάνω υπολογισμό για διάφορες τιμές του planewavecutoff (300, 330, 360, ... eV). Πόση είναι η τιμή του για να βρείτε την ενέργεια με ακρίβεια πρώτου δεκαδικού ψηφίου;
- Προσθέστε τις παρακάτω εντολές στο πρόγραμμά σας για να βρείτε το μήκος του δεσμού στο CO (δείτε και το παράδειγμα). Αλλάξτε και τα ονόματα των αρχείων στον ορισμό του calc.
calc.SaveMemory(True) calc.StayAliveOn() from ASE.Dynamics.QuasiNewton import QuasiNewton dyn = QuasiNewton(atoms, fmax=0.05) dyn.Converge() print "atom positions, energy" print atoms.GetCartesianPositions(), atoms.GetPotentialEnergy()
-
Τροποποιήστε κατάλληλα το πρόγραμμα plotwavefunction για να δείτε τις κυματοσυναρτήσεις στο μόριο CO, px:
python -i CO_wavefunction.py 2
Βεβαιωθείτε ότι ταιριάζουν με αυτές που προβλέπει η θεωρία μοριακών τροχιακών. Δείτε και το πρόγραμμα. - Επαναλάβετε τους υπολογισμούς και για άλλα μόρια της επιλογής σας, (πχ O2, HCl, NH3, ...). Χρησιμοποιήστε τις ίδιες υπολογιστικές παραμέτρους που βρήκατε για το CO. Βρείτε τα μοριακά τροχιακά.
- Xρησιμοποιήστε τα αποτελέσματά σας για να υπολογίσετε αν είναι ενδόθερμες ή εξώθερμες απλές αντιδράσεις (πχ η H2+½O2→H2O). Υπολογίστε την ενθαλπία της αντίδρασης (H=U+PV) σε ΚΣ, θεωρώντας ότι έχουμε τέλειο αέριο. Περιγράψτε πώς θα υπολογίζατε την ελεύθερη ενέργεια της αντίδρασης (G=H-TS).
- Ακολουθήστε την άσκηση για να υπολογίσετε την πλεγματική σταθερά και το μέτρο ελαστικότητας του Al.
- Επαναλάβετε τον υπολογισμό για μια υποθετική δομή bcc του Al, και βρείτε τη διαφορά ενέργειας μεταξύ των δυο δομών.
- Εξηγείστε τι είναι η LDA και τι η GGA. Βρείτε πώς επηρεάζεται η παραπάνω διαφορά από τις διαφορετικές προσεγγίσεις στο δυναμικό ανταλλαγής και συσχέτισης (βάλτε στη συνάρτηση Dacapo όρισμα xc='LDA' ή 'PBE'. default είναι το Pw91). Βρείτε ποια προσέγγιση δίνει καλύτερο αποτέλεσμα σε σχέση με το πείραμα (1.3 kJ/mol).
- Υπολογείστε πώς εξαρτώνται από τη συγκεντρωση χ οι ιδιότητες ενός από τα παρακάτω κράμματα:
- Al-Ni
- Al-Ti
- Au-Cu
- Fe-Pt (Pt-fcc, FePt3-L12, FePt2-C15, FePt-L11, Fe2Pt-C15, Fe3Pt-DO3, Fe-bcc, δείτε εδώ για τις δομές)
- Si-Ge (Si-A4, Si3Ge-DO3, SiGe-C20 με χ1=0 και χ2=¼, SiGe3-DO3, Ge-A4)
- Για το κράμα AB, βρείτε το planewavecutoff και τον αριθμό k-points ώστε να είστε σίγουροι για την ενέργεια στο πρώτο δεκαδικό ψηφίο. Στα άλλα κράμματα AχB1-χ να χρησιμοποιήσετε το ίδιο planewavecutoff και αντιστρόφως ανάλογα k-points (L*n=L'*n'), χωρίς να αλλάξετε από άρτιο σε περιττό αριθμό ή το αντίστροφο.
- Για κάθε ένα από τα κράματα, ακολουθήστε το παραπάνω παράδειγμα του Al για να βρείτε τις πλεγματικές σταθερές. Σε περιπτώσεις που οι παράμετροι είναι πολλές, κρατήστε τους λόγους σταθερούς και μεταβάλλετε μόνο μία από αυτές.
- Βρείτε την πυκνότητα (σε gr/cc), το μέτρο ελαστικότητας (σε GPa), τις αποστάσεις μεταξύ πρώτων γειτόνων, την ενθαλπία σύνθεσης (χA+(1-χ)B→AχB1-χ+ΔΗ) και ό,τι άλλο θέλετε.
- Δώστε γραφικές παραστάσεις όπου χρειάζεται. Σχολιάστε τυχούσες αποκλίσεις από τον νόμο του Vegard, σύμφωνα με τον οποίο οι ιδιότητες των κραμμάτων AχB1-χ είναι γραμμικές συναρτήσεις του χ. Συγκρίνετε αν θέλετε με τη βάση δεδομένων του CAMD.
- Χρησιμοποιείστε τα προγράμματα Build.py και Al100.py. Α Υπολογίστε την επιφανειακή τάση με γραφική παράσταση της ενέργειας σαν συνάρτηση του Ν. Βάλτε το πρόγραμμα στο background:
qsub Al100.py
ήpython Al100.py &
Όσο τρέχει το πρόγραμμα (20-30 λεπτά), τρέξτε το Build.py και βεβαιωθείτε ότι πρόκειται για σωστή δομή. - Προσθέστε στην βιβλιοθήκη Build.py μια συνάρτηση FCC111. Φτιάξτε ένα πρόγραμμα Al111.py που να την χρησιμοποιεί στα πρότυπα του Al100.py.
- Υπολογίστε τις ενέργειες προσρόφησης H και CO στην Al(111). Η ενέργεια προσρόφησης ορίζεται ως Eads=E(X+Al)-E(Al)-E(X). Για το Η πάρτε E(H)=0.5*E(H2). Προσέξτε ώστε όλοι οι υπολογισμοί να γίνουν με τις ίδιες παραμέτρους. Τρέξτε το co@al111.py που υπολογίζει την ενέργεια και τις συντεταγμένες CO προσροφημένου on top στην Al(111). Μετά τροποποιήστε το ώστε να προσομοιώσετε προσρόφηση Η σε θέση fcc hollow (δηλαδή εκεί όπου θα έμπαινε το επόμενο άτομο Al αν το υλικό συνεχιζόταν και πάνω από την επιφάνεια). Δώστε τα μήκη των δεσμών Al-H, Al-C και C-O και συγκρίνετε με το μόριο CO.
-
Bρείτε τί ποσοστό της επιφάνειας θα καλύπτεται από CO και τι από H σε διάφορες συνθήκες. Χρησιμοποιήστε το μοντέλο του
Langmuir:
Οι σταθερές χημικής ισορροπίας δίνονται από την K=K0exp(Eads/kT). Πάρτε K0=10-5. P είναι η συνολική πίεση, ενώ PCO και PH2 είναι οι μερικές πιέσεις. Θεωρήστε ότι PH2+PCO=P. To θ συμβολίζει το ποσοστό της επιφάνειας που καλύπτεται από μόρια CO ή H ή είναι κενό (*). Ενδιαφέρουν ιδιαίτερα συνθήκες αποθήκευσης υδρογόνου (λιγότερο από 1 ppm CO, χαμηλή θερμοκρασία). Δώστε τις γραφικές παραστάσεις των θ σαν συνάρτηση της συγκέντρωσης CO και σαν συνάρτηση της θερμοκρασίας. Δείτε το coverage.py.H2+2*↔2H* (1) (PH2/P) θ*θ*=K1θHθH CO+*↔CO* (2) (PCO/P) θ*=K2θCO
Σε αυτό το εργαστήριο θα δούμε χαρακτηριστικές τεχνικές μελέτης της ηλεκτρονικής δομής ενός υλικού: την προσομοίωση σαρωτικού μικροσκοπίου σήραγγας (STM), τον υπολογισμό της δομής ζωνών (band structure) και την πυκνότητα καταστάσεων. Θα χρειαστούμε τα αρχεία
Al-fcc-single.nc al111_3.nc h@al111_3.nc co@al111_3.ncαπό τα εργαστήρια 2 και 3.
- Περιγράψτε σύντομα την αρχή λειτουργίας του STM. Χρησιμοποιήστε το stm.py για να δείτε προσομοίωση STM από την Al(111).
- Πάρτε τρεις εικόνες STM: Τοποθετήστε στην επιφάνειά σας ένα άτομο O σε θέση fcc hollow, με Δz=0.7 Α, όπως παρατηρείται πειραματικά στο άρθρο. Προσομοιώστε την εικόνα STM και συγκρίνετε με το πείραμα (εικ. 2 και 5). Επαναλάβετε τον υπολογισμό με Η στη θέση του Ο. Τι παρατηρείτε;
- Χρησιμοποιήστε την βιβλιοθήκη bstr.py και το πρόγραμμα al_bstr.py για να υπολογίσετε τη δομή ζώνης του Al κατά μήκος των γραμμών WΓ και ΓX. Προσθέστε τις κατευθύνσεις LK, KW, XW και συγκρίνετε με το αποτέλεσμα του παραπάνω αρχείου.
- Σχεδιάστε την πυκνότητα καταστάσεων του Al με το dos.py. Κορυφές στην πυκνότητα καταστάσων συδυάζονται με σχετικά επίπεδες μπάντες, ενώ τα ελάχιστα της πυκνότητας καταστάσεων βρίσκονται εκεί όπου οι μπάντες είναι απότομες. Γιατί; Ταυτοποιήστε τέτοια σημεία. Μοιάζει το αποτέλεσμα με την θεωρία ελευθέρων ηλεκτρονίων;
- Χρησιμοποιήστε το al_wavefunction.py για να σχεδιάσετε την κυματοσυνάρτηση της χαμηλότερης κατάστασης στο Al. Πώς μοιάζει; Πώς μοιάζουν οι επόμενες (για k=0); ξεχωρίζουν οι αντιδεσμικές;
- Υπολογίστε την δομή ζώνης στο Si (LΓXUKΓ) και συγκρίνετε με άλλους υπολογισμούς(άσκηση και εδώ.). Υπολογίστε την πυκνότητα καταστάσεων και σχολιάστε τη σχέση της με τη δομή ζώνης. Σχεδιάστε τις κυματοσυναρτήσεις και συγκρίνετε με την θεωρία LCAO
- H DFT μπορεί να χρησιμοποιηθεί για τον υπολογισμό των συχνοτήτων δόνησης των μορίων. Υπολογίστε τη δονητική συχνότητα ταλάντωσης τού διατομικού μορίου CO από την σταθερά δύναμης χρησιμοποιώντας τη διαφορά ενέργειας για μικρές απομακρύνσεις από τη θέση ισορροπίας. Θα χρειαστεί να χρησιμοποιήσετε το αποτέλεσμα της άσκησης (1.4). Συγκρίνετε με την πειραματική τιμή και σχολιάστε τυχόν αποκλίσεις. Γιατί τα διατομικά μόρια έχουν ένα κανονικό τρόπο ταλάντωσης (stretching mode); Ποιό είναι το φυσικό νόημα της συχνότητας που υπολογίζετε;
- Χρησιμοποιείστε το πρόγραμμα CO_vibrations.py για να βρείτε την δονητική συχνότητα του CO. Συγκρίνετε με την πειραματική τιμή.
- Επαναλάβετε τις (1) και (2) για άλλα διατομικά μόρια, όπως για παράδειγμα τα NO, HF, HCl, HBr.
- Γράψτε πρόγραμμα στο οποίο να τοποθετείτε ένα μόριο νερού σε κυβικό κουτί (π.χ. ακμής 6 Angstrom). Μπορείτε να χρησιμοποιήσετε σαν αρχικές συντεταγμένες των ατόμων (0,0,0) για το Ο και (-0.77,0,-0.6), (0.77,0,-0.6) για τα άτομα Η. Βρείτε τις θέσεις ισορροπίας όπως κάνατε για το μόριο CO. Στη συνέχεια, εκτελέστε το πρόγραμμα H2O_vib.py. Πόσες δονητικές συχνότητες βρίσκετε και γιατί; Συγκρίνετε τις τιμές των συχνοτήτων με τις πειραματικές τιμές και σχολιάστε τα αίτια πιθανών αποκλίσεων.
- Για να δείτε πώς κινούνται τα άτομα με τους κανονικούς τρόπους ταλάντωσης,
χρησιμοποιείστε την εντολή plottrajectory και τα αρχεία H2O_vib_mode_x.traj
(όπου x είναι ο αριθμός του τρόπου ταλάντωσης με την σειρά που τυπώνεται η
συχνότητά του). Χρησιμοποιώντας VMD μπορείτε να δείτε τις κινήσεις των ατόμων
σε μορφή "ταινίας". Αφού πάρετε τις πληροφορίες σε αρχεία .xyz με την εντολή
trajectory2xyz [traj filename] [xyz filename]
Για να δείτε την κίνηση του μορίου, δώστεvmd filename.xyz
- Επαναλάβετε τις (4) και (5) για άλλα πολυατομικά μόρια, όπως π.χ. NH3, CO2, ONF, HCN, OCS, CH4.
- Η DFT μπορεί να περιγράψει τον μαγνητισμό των ηλεκτρονίων στα στερεά. Η μέθοδος Spin-polarized Density Functional Theory, εκτός από τον ποσοτικό καθορισμό μαγνητικών ροπών σπιν, προσφέρει και στην κατανόηση των βασικών μηχανισμών μαγνητισμού στα στερεά. Στην άσκηση αυτή μελετάμε μεταβατικά μέταλλα (Fe, Co, Ni) σαν παραδείγματα στα οποία η DFT προβλέπει και ερμηνεύει μαγνητικές ιδιότητες. Ακολουθείστε την άσκηση για να μελετήσετε τον Fe (δομή bcc).
- Επαναλάβετε για άλλα μέταλλα.
- Σε αυτό το εργαστήριο μελετάμε την κίνηση ατόμου προσροφημένου σε επιφάνεια. Η διάχυση ατόμων σε επιφάνειες, εκτός από το θεμελιώδες της ενδιαφέρον, σχετίζεται με φαινόμενα και διεργασίες μεγάλης τεχνολογικής σημασίας, όπως είναι η μοριακή επιταξία και η κατάλυση. Ακολουθήστε την άσκηση για να μελετήσετε τους μηχανισμούς αυτο-διάχυσης πάνω στην επιφάνεια Al(110). Βρέστε τις διαδρομές ελάχιστης ενέργειας και τις μεταβατικές καταστάσεις με την μέθοδο "Nudged Elastic Band" για τις τρεις διεργασίες που περιγράφονται στην άσκηση και συγκρίνετε τα αντίστοιχα φράγματα ενέργειας.
-
Για τον υπολογισμό του ρυθμού αναπήδησης (hopping rate) των ατόμων, k ,
από ένα σημείο ελάχιστης ενέργειας της επιφάνειας σε άλλο
χρησιμοποιείται η θεωρία μεταβατικής κατάστασης (transition state
theory), που στην απλούστερη αρμονική προσέγγιση δίνει
k=ν0e-Ea/kBT. όπου Ea είναι το φράγμα ενέργειας και ν0 είναι η συχνότητα ταλάντωσης του προσροφημένου ατόμου γύρω από το σημείο ισορροπίας (ελάχιστης ενέργειας). Χρησιμοποιώντας την παραπάνω σχέση, σχεδιάστε την γραφική παράσταση k vs 1/kBT για τις διεργασίες που μελετήσατε παραπάνω. -
Ακολουθήστε την άσκηση
που προσεγγίζει (αναλυτικά) την διάχυση ατόμου υδρογόνου προσροφημένου σε
μεταλλική επιφάνεια. Για τον υπολογισμό του ρυθμού αναπήδησης χρησιμοποιήστε
το αποτέλεσμα της αρμονικής θεωρίας μεταβατικής κατάστασης,